|
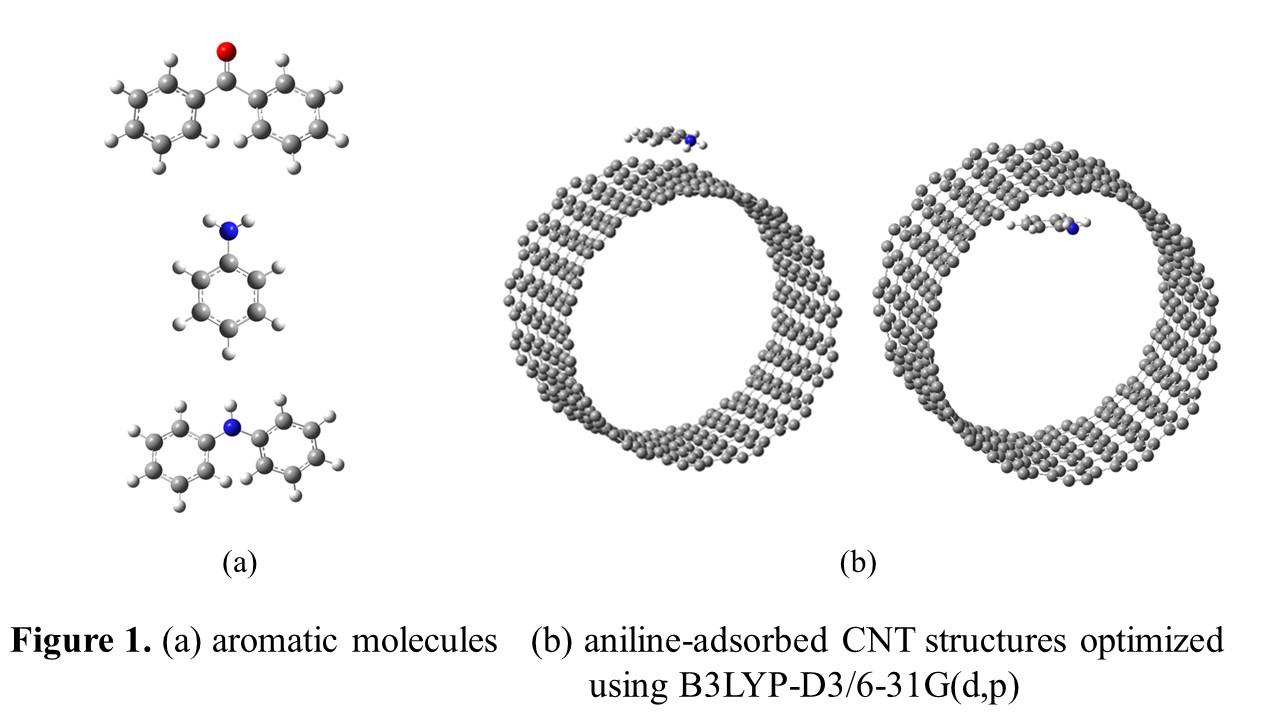
In this research, we have studied, by performing quantum chemical calculations, to find inter-molecular bonding strength between carbon nanotubes (CNT) and such molecules as aniline, benzophenone, and diphenylamine and favorable binding site on the CNT surfaces. Quantum chemical calculations were performed using B3LYP-D3/6-31G(d,p), which has shown high performances on intermolecular weak interaction calculations. We made the model of CNT structure (diameter: 1.97 nm) by cutting off to be an appropriate length and made each of the aromatic molecules adsorbed to the inside and outside of the CNT structure, performed the geometry optimization calculations and obtained the predicted binding energies. As the lengths of the CNT structure became longer, their binding energies increased slightly, but we found that their binding energies were converged at some predictable limit. The results of all the calculations were compared with the experimental data and it was confirmed that the quantum chemical calculations can explain the chemical phenomena observed in the experiment reasonably. |
|
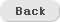
;)