|
|
| Type |
Oral Presentation |
| Area |
Oral Presentation for Young Scholars in Physical Chemistry |
| Room No. |
Room 208+209+210 |
| Time |
THU 10:00-: |
| Code |
PHYS.O-6 |
| Subject |
Target-based drug discovery through inverse quantitative structure-activity-lipophilicity relationships and molecular simulation |
| Authors |
Petar Zuvela, Jay Liu1,*, Myunggi Yi2, Paweł Pomastowski3, Gulyaim Sagandykova4, Tomasz Bączek5, Jarosław Sławiński6, Ming Wah Wong, Bogusław Buszewski3
Department of Chemistry, National University of Singapore, Singapore
1Department of Chemical Engineering, Pukyong National University, Korea
2Department of Biomedical Engineering, Pukyong National University, Korea
3Department of Environmental Chemistry and Bioanalytics, Nicolaus Copernicus University, Poland
4Nicolaus Copernicus University, Department of Environmental Chemistry and Bioanalytics, Poland
5Department of Pharmaceutical Chemistry, Medical University of Gdańsk, Poland
6Department of Organic Chemistry, Medical University of Gdańsk, Poland
|
| Abstract |
|
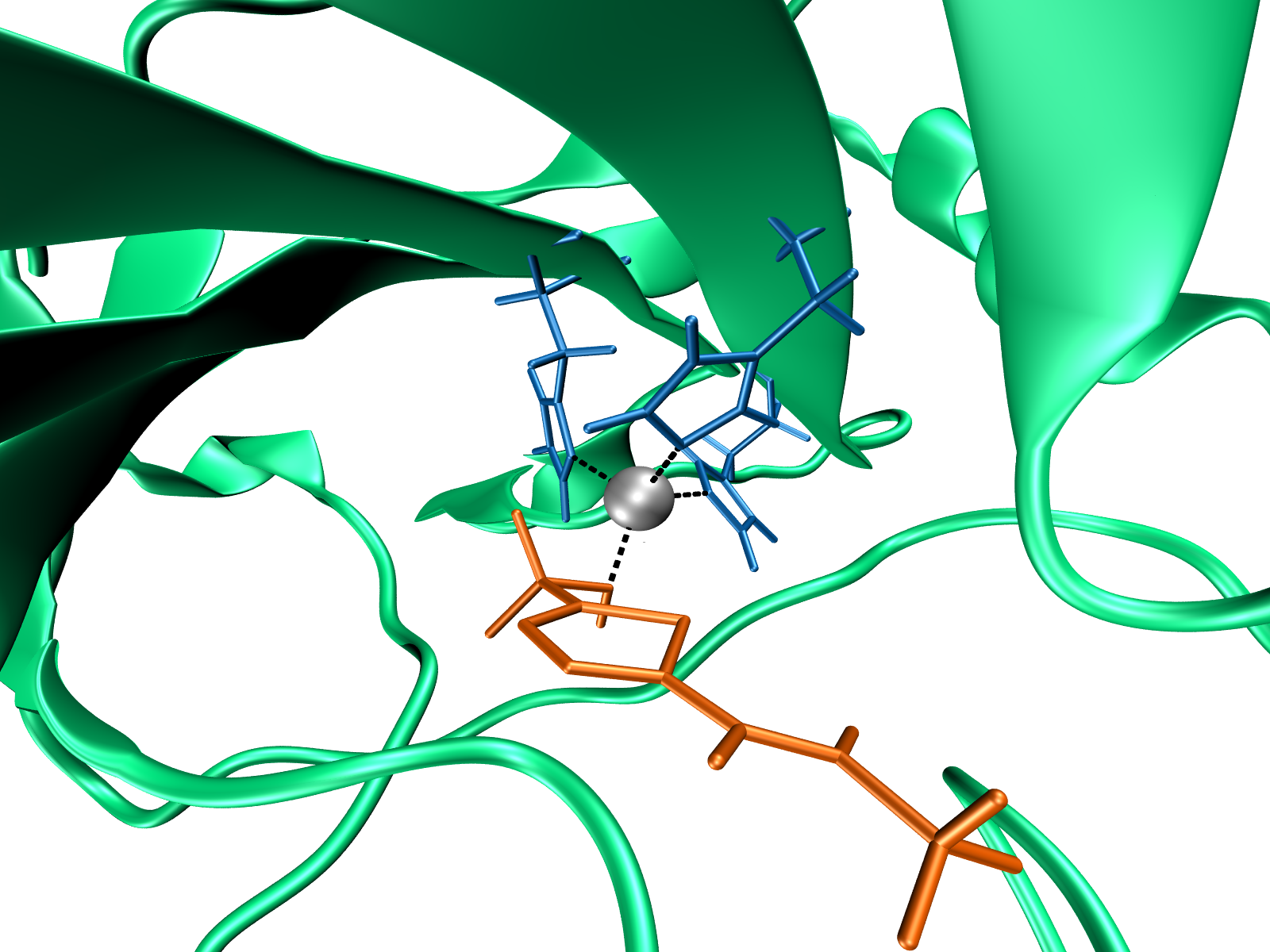
If drug properties are a function of its molecular structure, then the ideal drug candidate’s molecular structure is an inverse function of their maximum. This is the reasoning behind a drug discovery methodology presented in this work. As a model system, 45 sulfonamide derivatives in complex with carbonic anhydrase IX (CA IX) were used for development of strongly predictive quantitative structure-activity-lipophilicity relationships (QSALR) model. The model was reversed through numerical optimization in order to obtain an optimal molecular structure, in respect to a reference sulphonamide. Simultaneously, eight testing ligands were complexed with CA IX and characterized experimentally. Gel electrophoresis and MALDI-TOF/TOF-MS were used to determine the mass of the enzyme and explore the dimerization of CA IX. RP-LC and stopped-flow spectrophotometry were used for determination of lipophilicity and inhibitory activity, respectively. FTIR was used to elucidate the structural changes between the recombinant CA IX and its complexes. Molecular dynamics (MD) simulations were performed to complement the experiments, and vice-versa. Results have shown that upon simulation of acetazolamide in complex with CA IX, the system conserved the experimental flexibility profile, and the average radius of gyration obtained by MD analysis agreed well with its empirical value. MALDI-TOF-TOF/MS has confirmed strong binding of inhibitors to CA IX, while subtle weaker interactions between the ligands and CA IX were elucidated with spectroscopic experiments and MD analysis. |
|
|
| E-mail |
petar.zuvela@nus.edu.sg |
|
;)